
什么是 SimpleM?
在 GWAS(全基因组关联研究)中,我们通常需要对数百万个单核苷酸多态性(SNPs)进行假设检验,从而发现与目标表型相关的遗传变异。然而,这种大规模的多重检验问题容易导致假阳性,因此需要对显著性水平进行矫正(如 Bonferroni 矫正)。
然而,基因组数据中的 SNPs 并非完全独立,它们通常呈现高度的连锁不平衡(Linkage Disequilibrium, LD)。因此,传统的多重检验方法可能会过于严格,导致我们错失真正的信号。
SimpleM 是一种用于计算 “有效检验数量”(Effective Number of Tests, Meff) 的统计方法。通过对遗传数据中 LD 矩阵的特征值分解(Eigenvalue Decomposition, EVD),SimpleM 能够估计 SNPs 的独立信息成分数量,从而提供一种合理的多重检验矫正标准。
有效检验数量(Meff)
有效检验数量的核心思想是通过度量数据的相关性,简化假设检验的规模:
1. 直观解释:
Meff 是一个反映 SNP 独立性的信息量指标。对于高度相关的 SNPs(如位于同一个 LD 区域的变异),它们共享的大部分信息可以被归纳为一个“独立单元”。因此,实际需要检验的有效独立单元比总 SNP 数量要少。
2. 数学定义:
在 SimpleM 方法中,基于 LD 矩阵的特征值 (\lambda_i) 分解,Meff 的公式为:

其中, lambda i是 LD 矩阵的特征值。
3. 应用场景:
Meff 被用来调整显著性阈值,比如替代传统 Bonferroni 矫正中的显著性水平:
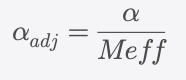
这比直接使用 SNP 总数更合理,也能提高研究的统计效能。
代码示例
首先需要说明的是,本文提供的代码并非100%准确,仅仅为各位朋友提供参考信息。请各位在使用前仔细阅读斟酌。首先是SimpleM的R代码,也是计算的核心脚本。
#============================================================================
# simpleM_Ex.R
#============================================================================
# License: GPL version 2 or newer.
# NO warranty.
#============================================================================
# citation:
#
# Gao X, Starmer J and Martin ER (2008) A Multiple Testing Correction Method for
# Genetic Association Studies Using Correlated Single Nucleotide Polymorphisms.
# Genetic Epidemiology 32:361-369
#
# Gao X, Becker LC, Becker DM, Starmer J, Province MA (2009) Avoiding the high
# Bonferroni penalty in genome-wide association studies. Genetic Epidemiology
# (Epub ahead of print)
#============================================================================
# readme:
# example SNP file format:
# row => SNPs
# column => Unrelated individuals
# The data file should contain only POLYMORPHIC SNPs.
# Missing values should be imputed.
# There should be NO missing values in the SNP data file.
# SNPs are coded as 0, 1 and 2 for the number of reference alleles.
# SNPs are separated by one-character spaces.
# You may need to change file path (search for "fn_In" variable)
# depending on where your snp file is stored at.
#============================================================================
# Meff through the PCA approach
# use a part of the eigen values according to how much percent they contribute
# to the total variation
Meff_PCA <- function(eigenValues, percentCut){
totalEigenValues <- sum(eigenValues)
myCut <- percentCut*totalEigenValues
num_Eigens <- length(eigenValues)
myEigenSum <- 0
index_Eigen <- 0
for(i in 1:num_Eigens){
if(myEigenSum <= myCut){
myEigenSum <- myEigenSum + eigenValues[i]
index_Eigen <- i
}
else{
break
}
}
return(index_Eigen)
}
#============================================================================
# infer the cutoff => Meff
inferCutoff <- function(dt_My){
CLD <- cor(dt_My)
eigen_My <- eigen(CLD)
# PCA approach
eigenValues_dt <- abs(eigen_My$values)
Meff_PCA_gao <- Meff_PCA(eigenValues_dt, PCA_cutoff)
return(Meff_PCA_gao)
}
#============================================================================
PCA_cutoff <- 0.995
#============================================================================
# fix length, simpleM
fn_In <- "请修改这里的文件路径/snpSample.txt"
mySNP_nonmissing <- read.table(fn_In, colClasses="integer")
numLoci <- length(mySNP_nonmissing[, 1])
simpleMeff <- NULL
fixLength <- 133
i <- 1
myStart <- 1
myStop <- 1
while(myStop < numLoci){
myDiff <- numLoci - myStop
if(myDiff <= fixLength) break
myStop <- myStart + i*fixLength - 1
snpInBlk <- t(mySNP_nonmissing[myStart:myStop, ])
MeffBlk <- inferCutoff(snpInBlk)
simpleMeff <- c(simpleMeff, MeffBlk)
myStart <- myStop+1
}
snpInBlk <- t(mySNP_nonmissing[myStart:numLoci, ])
MeffBlk <- inferCutoff(snpInBlk)
simpleMeff <- c(simpleMeff, MeffBlk)
cat("Total number of SNPs is: ", numLoci, "\n")
cat("Inferred Meff is: ", sum(simpleMeff), "\n")
#============================================================================
# end
下面是处理Raw数据生成矩阵并通过调用上面的simpleM_Ex.R 脚本进行计算的R代码
install.packages("data.table")
install.packages("corpcor")
# 加载必要的包
library(data.table)
library(corpcor)
# 读取基因型数据,使用plink获得的raw数据
genotype_data <- fread("修改为你的路径/my_genotype.raw", header=TRUE)
# 删除前6列非基因型信息(FID, IID, PAT, MAT, SEX, PHENOTYPE)
genotype_data <- genotype_data[, -(1:6), with=FALSE]
# 转置矩阵,使行表示SNP,列表示个体
genotype_matrix <- t(as.matrix(genotype_data))
# 检查基因型矩阵中是否有缺失值或无穷值
any(is.na(genotype_matrix)) # 返回TRUE则表示有NA值
any(is.infinite(genotype_matrix)) # 返回TRUE则表示有无穷值
# 使用平均数填充NA
#genotype_matrix[is.na(genotype_matrix)] <- rowMeans(genotype_matrix, na.rm = TRUE)
# 定义计算众数的函数
getmode <- function(v) {
uniqv <- unique(v[!is.na(v)]) # 去除NA后获取唯一值
uniqv[which.max(tabulate(match(v, uniqv)))] # 返回出现次数最多的值
}
# 使用众数替换每行中的缺失值
for (i in 1:nrow(genotype_matrix)) {
mode_value <- getmode(genotype_matrix[i, ])
genotype_matrix[i, is.na(genotype_matrix[i, ])] <- mode_value
}
# 检查是否还有缺失值
any(is.na(genotype_matrix)) # 如果返回FALSE,说明所有缺失值已被填充
# 将转置后的矩阵保存为纯文本文件,供SimpleM使用
write.table(genotype_matrix, file="将矩阵写入的文本文件/snpSample.txt", row.names=FALSE, col.names=FALSE, quote=FALSE, sep=" ")
# 在R中运行脚本
source("修改为你的simpleM_Ex.R文件的保存路径/simpleM_Ex.R")
在上面的示例代码中,对于缺失值的处理有很多方法,需要根据实际情况选择更合适的方案。这里仅供参考。处理后的结果,控制台会出现类似的信息:

由于我使用的芯片分型,因此原始SNP数量就很少。如果你使用的重测序数据Call的SNP,情况可能很不一样。同样,仅供参考。